### Séquençage ADN
Détermination de l'ordre d'enchaînement des nucléotides
d'un fragment d'ADN/ARN donné
### Pourquoi séquencer l'ADN?
Identification de gènes, de régions plus larges, de
chromosomes ou de génomes entiers
### Applications
- Biologie moléculaire
- Biologie évolutive
- Métagénomique
- Médecine
- Forensiques
### Un peu de bibliographie
- Revue sur l'histoire du début du séquençage et de la
bioinformatique : [*Hutchinson, Nucl Acids Res, 2007*](http://nar.oxfordjournals.org/content/35/18/6227.short)
- Revues sur les différentes méthodes de séquençage
- [*Metzker, Genome Res, 2005*](http://genome.cshlp.org/content/15/12/1767.short)
- [*Metzker, Nature Rev Genet, 2010*](http://www.nature.com/nrg/journal/v11/n1/abs/nrg2626.html)
- [*Schadt et al, Hum Mol Genet, 2010*](http://hmg.oxfordjournals.org/content/19/R2/R227.short)
### Avantages
- Haut-débit
- Coûts faibles
### Limites
- Amplification
- Risque d'erreur
- Augmentation de la complexité et du temps associé
à la préparation
- Génération importante de données
- Traitement
- Stockage
- Séquences courtes
## Entre la 2e et 3e génération
### Ion Torrent
- Avantages
- Pas besoin de lumière, scanning et caméras
- Limites
- Système "wash-and-scan" avec une amplification PCR
### Helicos Genetic Analysis Platform
Imagerie d'ADN individuels fixés sur une surface plane
pendant leur élongation avec une polymérase modifiée et
des nucléotides fluorescents
### Helicos Genetic Analysis Platform
- Avantages
- Pas de PCR
- Limites
- Arrêt de l'élongation
- Taux d'erreur > 5%
### Troisième génération
3 principales méthodes
- SBS
- Nanopores
- Imagerie directe
### Sequencing by synthesis (SBS)
2 techniques
- Séquençage en temps réel de molécules simples
- Séquençage en temps réel avec observation du transfert
d'énergie entre molécules fluorescentes
### Troisième génération
Avantages
- Pas besoin de PCR (molécule simple)
- Haut débit
- Délai de production plus courts
- Reads plus longs
- Plus forte précision
- Besoins en ADN plus faibles
- Coûts plus faibles
## Comparaison des générations et des méthodes
### Choix de la plateforme
A prendre en considération
- Application souhaitée
- Coût d'un run et de son traitement
- Longueur des reads
- Nombre de reads par run
- Taux d'erreur
- Disponibilité
### Longueur des reads
- Reads longs
- Facilité d'assemblage
- Optimal pour des génomes jamais séquencés et la caractérisation du transcriptome
- Reads courts
- Faibles coûts
- Plus forte couverture
- Re-séquençage pour applications basées sur la fréquence (comptage)
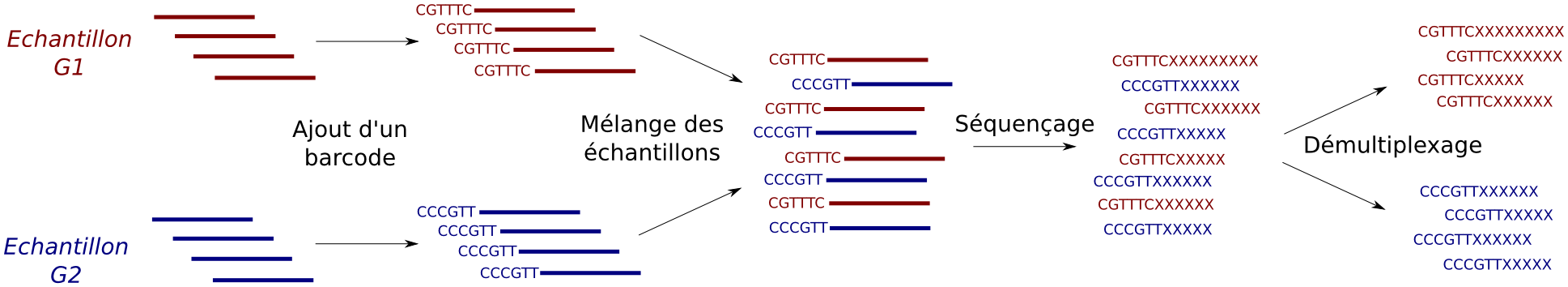
### Multiplexage
Moyen de diminuer les coûts de séquençage
- Ajout d'un tag d'identification pour chaque échantilon
- Mélange des échantillons
- Préparation et séquençage en parallèle
- Tri des séquences en fonction des échantillons
(information de source contenue dans la séquence)
# Traitement des données issues du séquençage
### Traitement des données issues du séquençage
- Gestion des données
- Prétraitement des données
- Contrôle de la qualité des séquences
- Assemblage
- Analyses des séquences
### Types de données
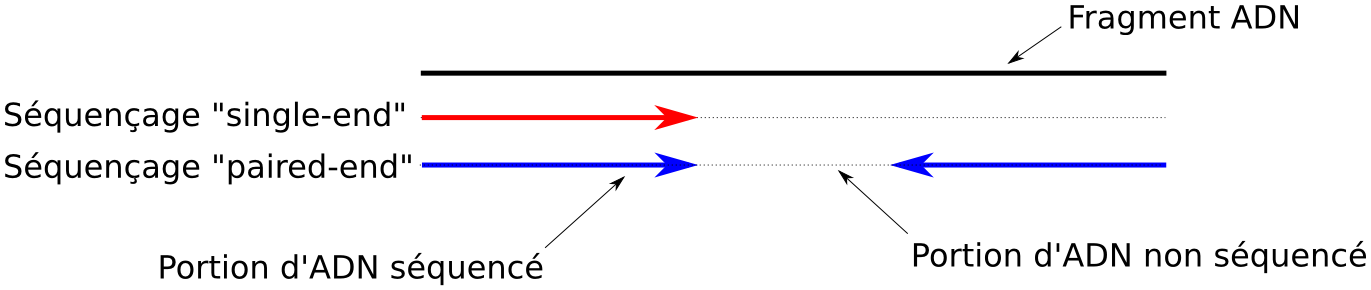
### Types de données
- Séquençage single-end
- 1 fichier de sortie
- Séquençage paired-end
- 2 fichiers de sortie
- Concaténation des fichiers en faisant attention au sens de lecture
### Formats des données brutes
- FastA: Format répandu de stockage des séquences biologiques
- FastQ (*Illumina*)
- SFF (*Roche 454*)
- SRF (*Helicos*)
- HDF5 (*PacBio, Applied Biosystems, Oxford Nanopore*)
### FastA
```
> Identifiant (Commentaire)
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
> Identifiant2 (Commentaire)
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
XX
```
### FastQ
```
> Identifiant (Commentaire)
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
+
QQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQ
> Identifiant2 (Commentaire)
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
+
QQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQ
```
### Score de qualité
Mesure de qualité de l'identification des bases par le
séquençage ADN
Score assigné à chaque base dans les sorties des séquenceurs
### Score de qualité

### Codage du score de qualité

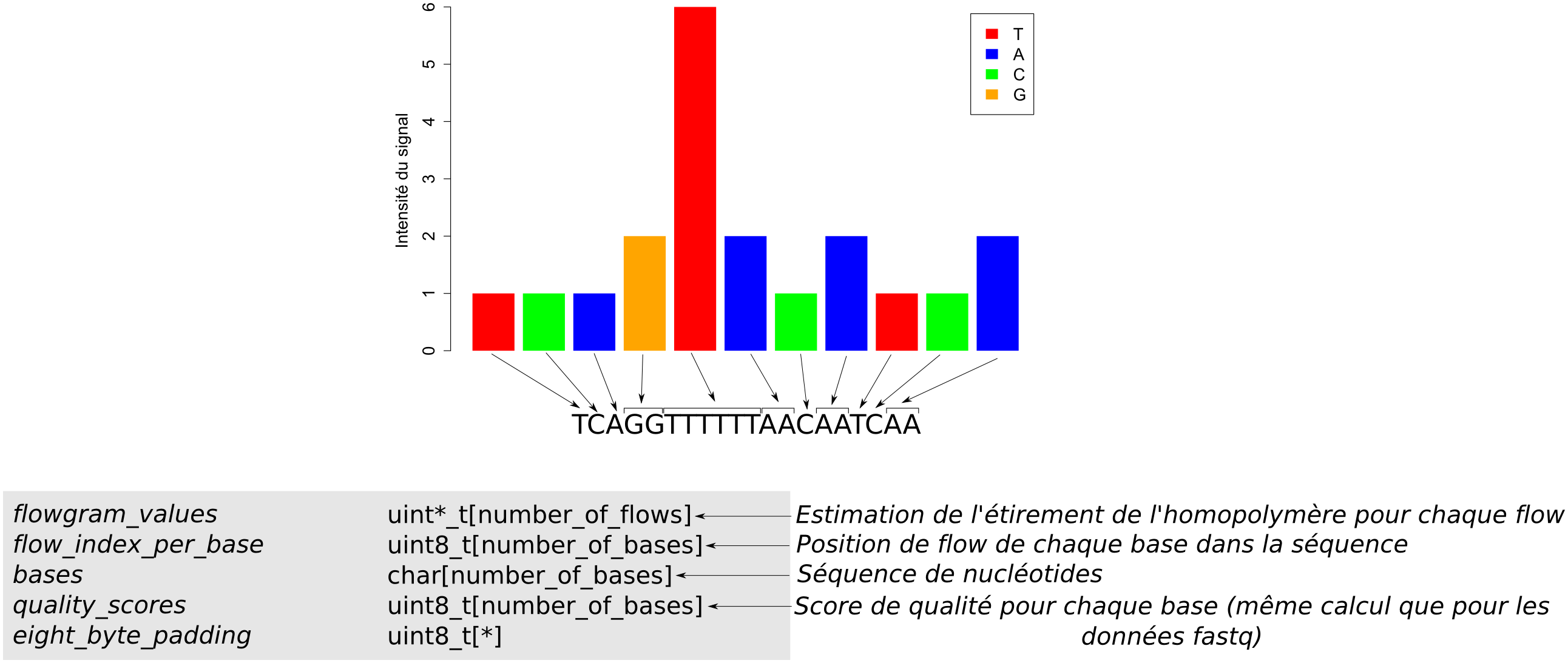
### Standard Flowgram Format (SFF)
- Format de sortie de 454 et Ion Torrent
- Fichier binaire
- Section en-tête commune à tous les reads
- Pour chaque read
- Section en-tête du read
- Section des données du read
### En-tête globale d'un fichier SFF

### En-tête pour un read dans un fichier SFF

### Données d'un read dans un fichier SFF
## Prétraitement des données
### Primers, adaptateurs, barcodes

### Multiplexage / Démultiplexage
## Contrôle de la qualité des séquences
### Contrôle de la qualité des séquences
Plusieurs paramètres à vérifier
- Longueur des séquences
- Score de qualité des bases
- Contenu en bases
- Duplications de séquence
- Complexité des séquences
- Contamination
### Score de qualité des bases

### Score de qualité des bases
2 niveaux à vérifier
- Score de qualité de la séquence entière
- Eliminer les séquences avec score moyen < 25
- [Huse et al, Genome Biology, 2007](http://link.springer.com/article/10.1186/gb-2007-8-7-r143)
- Score de qualité par base
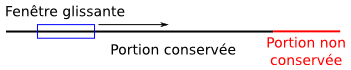
### Coupure des séquences basées sur le score de qualité par base
Coupure de longueur fixe

### Coupure des séquences basées sur le score de qualité par base
Coupure de longueur variable
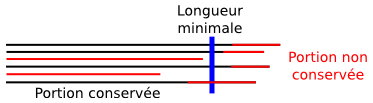
Utilisation d'une fenêtre glissante
### Exemples de contrôle de la qualité
- [Jeu de données utilisé en TP](data/correct_quality_dataset/SRR029695_fastqc.html)
- [Jeu de données de mauvaise qualité](data/bad_quality_dataset/SRR026762_fastqc.html)
### Assemblage
Alignement et fusion de reads en séquences ADN plus longues
### Assemblage
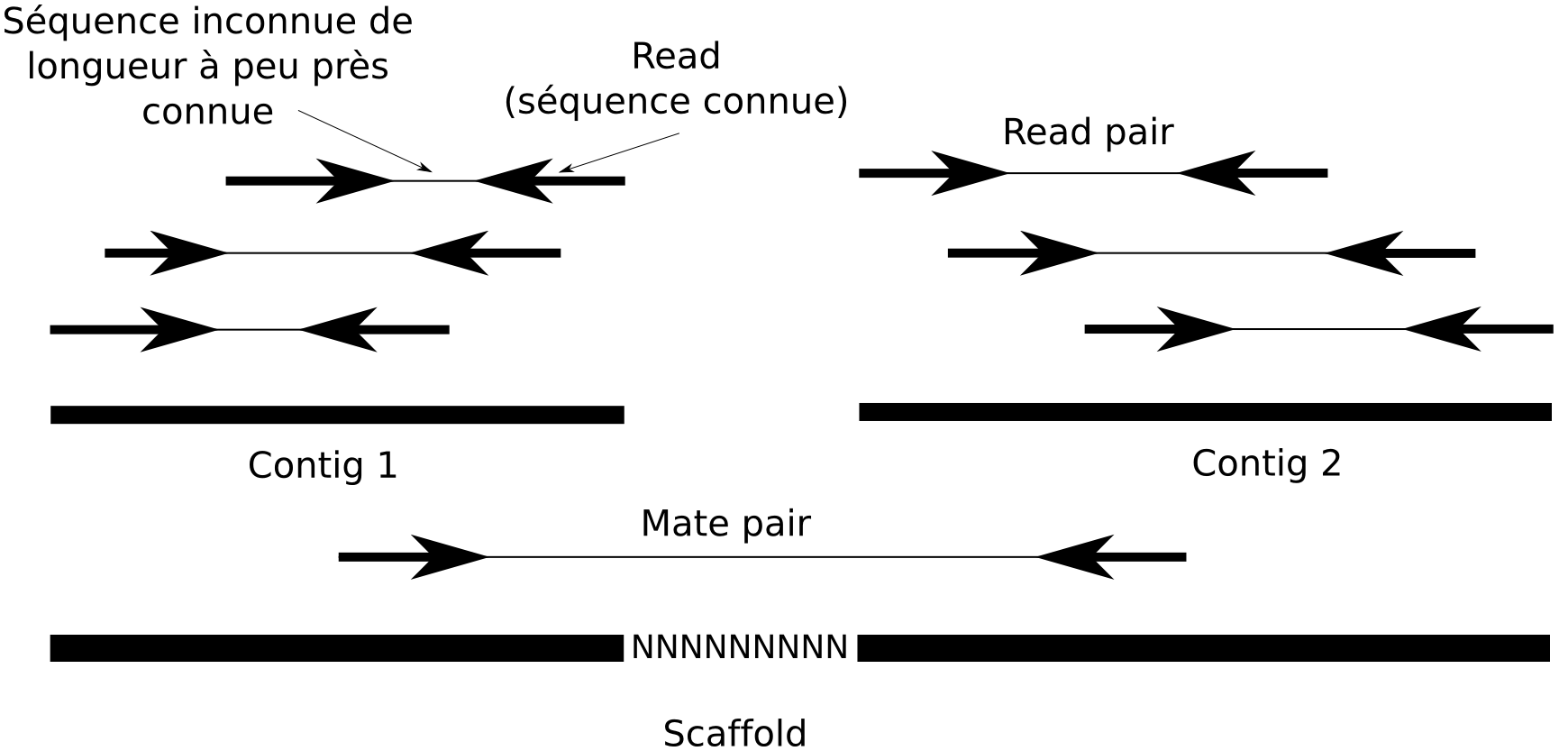
### Assemblage
- **Contig** : Séquence génomique continue et ordonnée générée par l'assemblage de reads qui se chevauchent
- **Scaffold** : Contigs chevauchant séparés par des gaps de longueur connue
### Statistiques de séquençage
- **Profondeur ou couverture** : Nombre moyen de fois qu'un nucléotide particulier est représenté dans une collection de reads aléatoire
- **Profondeur de couverture** : Nombre de reads x Longueur des reads / Taille de l'assemblage
### Métriques d'assemblage
Taille de l'assemblage
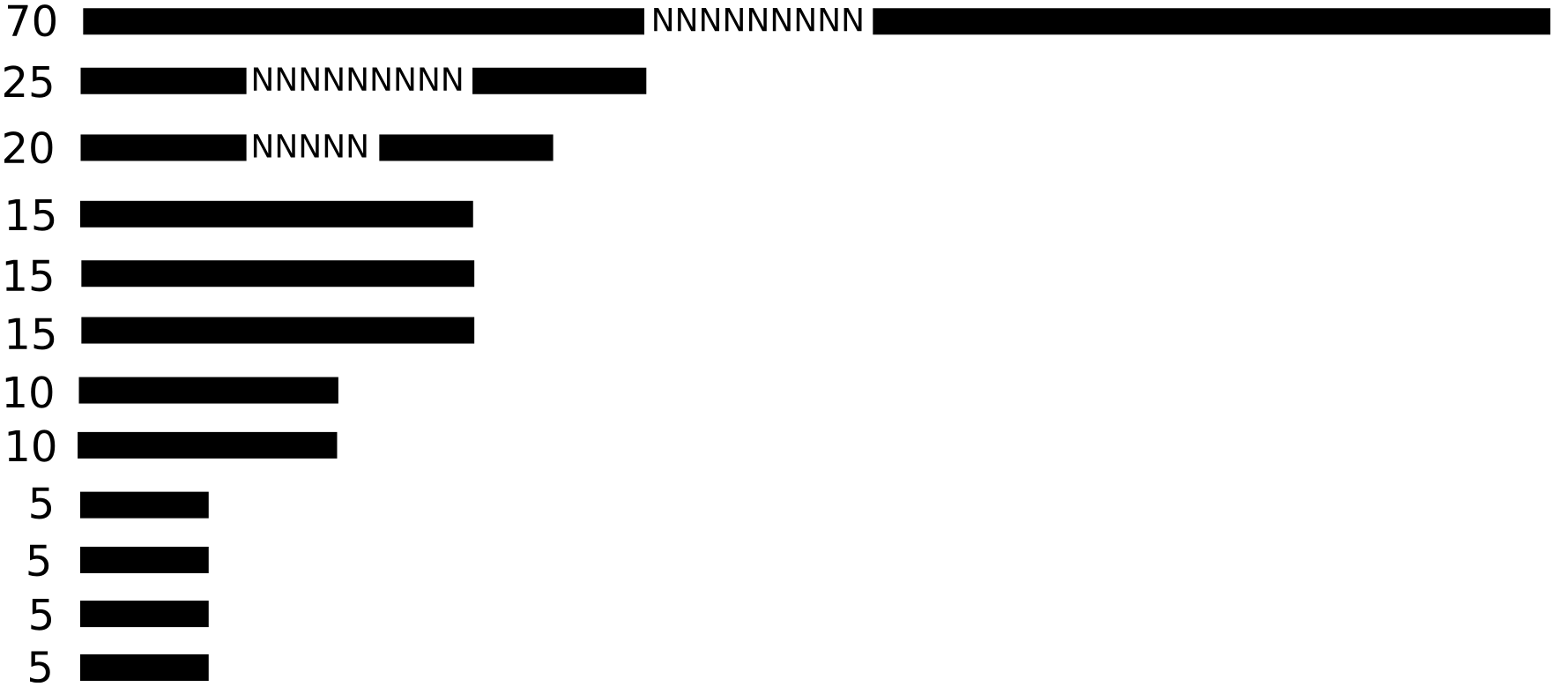
200 Mpb
### Métriques d'assemblage
Nombre de contigs/scaffolds

12
### Métriques d'assemblage
Taille moyenne des contigs/scaffolds

16.67
### Métriques d'assemblage
Nombre de N

23
### Métriques d'assemblage
N50

20
### Utilisation de l'assemblage
Alignement sur un génome de référence

- Identification de variants
- Reséquençage de génomes
### Utilisation de l'assemblage
Assemblage *de novo*
Construction des séquences ADN d'un organisme sans génome de référence
- Idéal
- Longs reads sans erreurs
- Problème de simple déduction
- Réalité
- Reads courts et sujets aux erreurs
- Problème d'inférence compliqué
### Outils d'assemblage
[> 75 outils](http://omictools.com/genome-assembly-category)
Note:
Choix populaire pour l'étude de l'expression des gènes
Alternative aux puces ADN pour l'identification de gènes
différentiellement exprimés
Note:
Analyses qualitatives et quantitatives des petits ARN non
codants
microRNAs and other regulatory small transcripts
## Références
- [Vidéo sur l'apport d'Ion Torrent par rapport à Illumina](https://www.youtube.com/watch?v=R8rEEUkfC9I)
- [QC fail](https://sequencing.qcfail.com)
- [Présentation de l'assemblage](http://fr.slideshare.net/kbradnam/assemblathon-2-talk?qid=31d4f62b-27c2-43d8-bf89-9f7074cedf67&v=&b=&from_search=1)
- [Différentes applications du séquençage haut-débit](http://omictools.com/sequencing-category)
## [Métagénomique](metagenomique.html)